Service de Pharmacologie Clinique
Présentation
Le service de Pharamcologie Clinique assure :
- le dosage des médicaments dans le cadre du suivi thérapeutique,
- les études de bioéquivalance,
- la pharmacogénétique,
- les expertises médico-légales,
- et la recherche clinique et expérimentale.
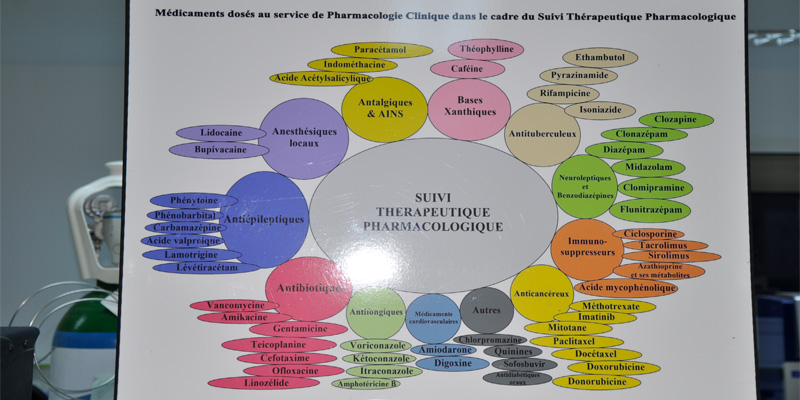
La liste des médicaments dosés ne cesse de s'allonger pour s'adapter aux besoins exprimés par les confrères cliniciens et suivre l'évolution scientifique.
Les modalités de prélèvements, le mode d'acheminement et l'interprétation des résultats sont propres à chaque situation clinique d'où l'obligation d'accompagner chaque demande de dosage par une fiche de renseignements spécifique pour chaque dosage téléchargeable à partir de ce lien
Présentation de la Bioéquivalence
Les études de bioéquivalence sont obligatoires avant la commercialisation de certain médicaments ou spécialités génériques.
La spécialité générique désigne toute spécialité pharmaceutique ayant la même forme galénique et la même composition qualitative et quantitative en principes actifs que la spécialité de référence, et dont la bioéquivalence avec cette dernière a été démontrée par des études de biodisponibilité appropriées et les nouvelles exigences d’enregistrement des médicaments génériques de la DPM et qui imposent des études de bioéquivalence sans possibilité d’exemption pour les formes pharmaceutiques nécessitant ces études selon le référentiel de l’OMS.
Les études de bioéquivalence constituent une garantie pour le prescripteur et le consommateur que les médicaments génériques disponibles sur le marché sont équivalents au produit princeps de référence (sécurité et efficacité).
D’après la loi n° 2008-32 du : 13 Mai 2008, modifiant et complétant la loi n°73-55 du 3 Août 1973 portant organisation des professions pharmaceutiques, la démonstration de la bioéquivalence de la spécialité générique avec la spécialité de référence s’établit par des études appropriées de biodisponibilité comparée. La dispense de ces études s’établit sur la base de critères scientifiques fixés par arrêté du Ministre de la Santé.
Ces études s’effectuent sur des volontaires sains. Mais, vu l’absence d’un cadre réglementaire pour la réalisation des essais cliniques, en Tunisie, un moratoire a été observé lors de la demande de l’Autorisation de Mise sur le Marché pour la commercialisation des spécialités génériques pour la majorité des produits où l’étude de bioéquivalence était nécessaire.
Actuellement et depuis la publication du décret autorisant l’expérimentation sur le volontaire sain en octobre 2014 ( Décret n° 2014-3657 du 3 octobre 2014, modifiant et complétant le décret n° 90-1401 du 3 septembre 1990), ces études sont devenues possibles en Tunisie.
L’inauguration de l’Unité de Bioéquivalence du Centre National « Chalbi belkahia » de pharmacovigilance a eu lieu le 11 Novembre 2019 par la ministre de santé Sonia BEN ECHIKH.
Questions concernant la Bioéquivalence
Quels sont les paramètres pharmacocinétiques mesurés lors d’une étude de bioéquivalence ?
Réponse :Au terme d’une étude de bioéquivalence, trois paramètres pharmacocinétiques sont mesurés : - L'aire sous la courbe de la concentration plasmatique en fonction du temps (ASC) qui mesure le taux d’absorption de la molécule. - La concentration maximale sanguine (Cmax). - Le délai pour atteindre la concentration sanguine maximale (Tmax). Suite à l’analyse des résultats de l’étude, deux formulations sont considérées bioéquivalentes, si les intervalles de confiance à 90?s rapports des paramètres pharmacocinétiques (ASC et Cmax) moyens (en échelle logarithmiques) sont inclus dans l’intervalle de confiance [80 %-125%].
Comment est démontrée la bioéquivalence d’un médicament générique par rapport à un médicament de référence ?
Réponse :Les études de bioéquivalence permettent de s’assurer que les deux médicaments ont la même biodisponibilité. Elle consiste à administrer alternativement le médicament de référence et le médicament générique au même sujet de façon croisée dans un ordre déterminé par tirage au sort selon un protocole d’administration bien défini (dose unique). Une période de « wash-out » est nécessaire entre les deux prises pour éliminer le médicament de l’organisme. L’essai est réalisé chez des sujets volontaires sains ayant consentis à participer à l’étude.
Qu’est ce qu’un médicament générique ?
Réponse :Le médicament générique est une spécialité pharmaceutique ayant la même forme pharmaceutique et la même composition qualitative et quantitative en principe actif, et dont la bioéquivalence avec la spécialité de référence a été démontrée par des études appropriées.
Les médicaments génériques sont soumis à une autorisation de mise sur le marché (AMM), qui nécessite la démonstration de la qualité pharmaceutique et de la bioéquivalence, c’est-à dire une équivalence de la biodisponibilité. Le médicament générique contient donc le même principe actif que le médicament de référence et obligatoirement en même quantité.
Pourquoi les médicaments génériques sont-ils moins chers que les médicaments de référence?
Réponse : Les médicaments génériques coutent généralement beaucoup moins cher que les médicaments de référence vu qu’ils ne font suite ni à une recherche ni à des essais cliniques.
Génériques
Présentation du suivi thérapeutique
Lors de la mise en route d'un traitement, il est nécessaire de chercher à obtenir le maximum d'efficacité et le minimum d'effets indésirables en adaptant le schéma thérapeutique aux besoins et à l'état général du patient.
Pour certains médicaments, cette adaptation peut se faire sur la base d'éléments cliniques et/ou biologiques.
Pour les autres, ces éléments sont insuffisants, et ne permettent pas d'assurer au malade le meilleur rapport bénéfice/risque. Dans ce cas, le dosage des médicaments trouve pleinement son indication et permet d'atteindre cet objectif.
Toutefois, ce dosage n'est possible que :
Pour les médicaments ayant une bonne corrélation entre les effets thérapeutiques et/ou indésirables (de mécanisme toxique) et les concentrations sanguines.
S'il existe une technique validée, fiable et facile à mettre en routine.
Habituellement, le dosage des médicaments permet de vérifier si la concentration sanguine est dans la zone d'efficacité thérapeutique ou intervalle thérapeutique. Ainsi, le dosage permet d’augmenter l’efficience des médicaments et d’assurer la sécurité des patients.
Le Service de Pharmacologie Clinique assure le dosage de plus de 10000 tests par an. Cette activité est indispensable et incontournable dans le suivi thérapeutique de certaines pathologies chroniques et/ou graves comme la greffe d’organes et de moelle osseuse, l’épilepsie, l’insuffisance rénale, le cancer, etc…
Le Service de Pharmacologie Clinique est considéré comme un service de référence et d’excellence dans le domaine du suivi thérapeutique pharmacologique de par le nombre de médicaments dosés, plus de 70 médicaments dosés en routine, de par le nombre de test réalisés, avec une base de données de dossiers malades informatisées de plus de 60 000 dossiers, crée en 2009, et de par le nombre de ses partenaires (une centaine de partenaire).
En effet, plusieurs conventions ont été signées dans le cadre de la coopération de notre Service avec de nombreuses structures Hospitalo-universitaires et de Recherche.
Cette coopération vise à atteindre les objectifs suivants :
Mutualiser les expertises dans le domaine du STP, de la pharmacocinétique et les bonnes pratiques.
Transfert des techniques par les études de pharmacocinétique mises au point vers les patients en collaboration avec nos partenaires cliniciens.
Renforcer et enrichir notre savoir-faire par des technologies innovantes.
Intervalles thérapeutiques des médicaments
Intervalles thérapeutiques des médicaments
| Classes pharmacologiques | Médicaments | Fourchettes thérapeutiques |
|---|---|---|
| IMMUNOSUPPRESSEURS | Ciclosporine* | C0 : 75-300 ng/ml, C2h : 500-1200 ng/ml Les c0 et c2h de la cyclosporine variant à l’intérieur de la fourchette thérapeutique en fonction de la nature de l’indiction, de la période de prescription et de l’état clinique et biologique du patient. |
| Tacrolimus* | Phase aigue (J0-J42) C0 : 10-15 ng/ml, Phase chronique (>J42) C0 :5-10 ng/ml |
|
| Sirolimus | C0 :4-12 ng/ml |
|
| Acide Mycophénolique* (AMP) |
Greffe Rénale C0 : 1,5-3 µg/ml, |
|
| Azathioprine (6TG) | C2h :230-450 pmol/8*108 GRC |
|
| ANTICANCEREUX | Méthotrexate | Leucémie : H24<5 µmol/l –H48<0,5 µmol/l – H72<0,05 µmol/l Ostéosarcome : H24<10 µmol/l –H48<1 µmol/l – H72<0,1 µmol/l |
| Imatinib | C0 :>1000 ng/ml |
|
| Mitotane | C0 :14-20 µg/ml |
|
| ANTIEPILEPTIQUES | Phénytoine | Adulte : C0 :10-20 µg/ml, Sujet âgé et enfant : C0 :5-15 µg/ml |
| Phénobarbital | Adulte : C0 :15-30 µg/ml, Enfant : C0 :20-40 µg/ml |
|
| Acide valproique | C0 :50-100 µg/ml |
|
| Carbamazépine | Monothérapie C0 :6-12 µg/ml |
|
| Thiopental | Anesthésie : 15-40 µg/ml |
|
| Lamotrigine | C0 :3-14µg/ml |
|
| Levetiracetam | C0 :10-40µg/ml |
|
| Felbamate | C0 :30-80µg/ml |
|
| ANTIFONGIQUE | C0 :1,5-5 µg/ml |
ANESTHESIQUES LOCAUX | Lidocaïne | C0 :2-5 µg/ml |
| Bupivacaïne | Seuil de toxicité > 1,5 µg/ml |
|
| Ropivacaïne | Interval de toxicité : 1,5-4 µg/ml |
ANTALGIQUES & ANTI-INFLAMMATOIRES NON STEROIDIENS | Paracétamol | Seuil de toxicité : H4 >200 µg/ml, H8 >100 µg/ml, H10 >60 µg/ml, H15>30 µg/ml |
| Acide acétylsalicylique | C0 :100-250 µg/ml (effet anti-inflammatoire), Toxicité >300 µg/ml |
ANTIBIOTIQUES | Vancomycine | C0 :10-12 µg/ml Cmax :20-40 µg/ml, PSE: 15-20 µg/ml |
| Amikacine | Dose fractionnées : C0 <5 µg/ml Cmax :20-30 µg/ml |
|
| Gentamicine | Dose fractionnées : C0 <2 µg/ml Cmax :5-8 µg/ml |
|
| Téicoplanine | C0 :10-40 µg/ml C1h :50-70 µg/ml |
|
| ANTITUBERCULEUX | Isoniazide | Test d’acétylation effectué à T3h : I3<0,6 :Acétyleur rapide, I3>0,6 :Acétyleur lent |
| Rifampicine | Activité antituberculeuse C2h:6-18 µg/ml |
MEDICAMENTS CARDIOVASCULAIRES | Digoxine | Enfant: 1-2,5 ng/ml, Adulte: 0,8-2 ng/ml |
| Amiodarone | C0 :0,5-2,5 µg/ml |
BASES XANTHIQUES | Théohylline | C0 :10-15 µg/ml |
| Caféine | C0 :8-20 µg/ml |
NEUROLEPTIQUES ET BENZODIAZEPINES | C0 :350-600 ng/ml |
| Clonazépam | C0 :15-50 ng/ml |
ANTIDIABETIQUES ORAUX *** | Glibenclamide | C0 :30-200 ng/ml |
| Glimépiride | C0 <300 ng/ml |
QUININES | Hydroxychloroquine | C0 >1000 ng/ml |
| Chloroquine | C0 :100-200 ng/ml toxicité >600 ng/ml |
* : Des cinétiques abrégées peuvent être faites pour ces 3 immunosuppresserus. Ciclosporine et Tacrolimus : prèlèvements à T0, T1h, et T3h.
AMP : T0, T30mn, et T90mn.
C0 : Concentration plasmatique résiduelle : Prélèvement juste avant la prise médicamenteuse.
Cnh : Prèlèvement n heures (s) après médicamenteuse.
PSE : Pousse Seringue Electrique.
** Des courbes de pharmacocinétique peuvent être réalisées à la demande en fonction de la situation clinique.
*** Le dosage peut être réalisé dans le cadre de l’exploration d’une hypoglycémie factice.
Expertises médico-légales
Le Service de Pharmacologie Clinique est la référence nationale en matière de Pharmacologie médicolégale. Il reçoit des demandes croissantes de dosage des médicaments moyennant des réquisitions.
Laboratoire de recherche en pharmacologie clinique et expérimentale
Les axes de recherche
Axe 1 : Personnalisation de la posologie médicamenteuse en fonction des propriétés pharmacocinétiques des médicaments et évaluation de l’impact de cette approche en pratique clinique.
Axe 2 : Apport des études pharmacogénétiques dans la personnalisation de la prescription médicamenteuse dans la population tunisienne.
Axe 3 : Valorisation des substances bioactives tunisiennes et application dans des modèles animaux de pathologies humaines.
Pharmacogénétique
La pharmacogénétique étudie l’influence du génotype sur la variabilité de la réponse à un traitement médicamenteux. Son rôle est d’identifier les polymorphismes génétiques responsables dans 20-95% de la variabilité interindividuelle de la réponse à certains médicaments.
*Les objectifs généraux de la pharmacogénétique :
Etudier les profils génétiques impliqués dans la réponse aux différentes classes médicamenteuses de la population Tunisienne;
Evaluer l’impact des polymorphismes génétiques sur la réponse thérapeutique dans cette population;
Personnaliser la pharmacothérapie en sélectionnant le médicament adéquat et la dose optimale pour chaque patient en fonction des profils génétiques identifiés;
Réduire les coûts de la santé dus aux effets indésirables toxiques et aux cas d’inefficacité (échec thérapeutique) par certains polymorphismes génétiques en personnalisant la prescription médicamenteuse;
Optimiser l’utilisation du médicament en fonction des polymorphismes génétiques afin d’assurer une meilleure efficience des médicaments en particulier pour les thérapeutiques innovatrices très coûteuses.
* Objectifs spécifiques
Notre activité a pour objectif d’identifier des polymorphismes génétiques. Le but étant d’adapter au mieux la dose optimale du médicament afin d’obtenir un meilleur bénéfice/risque.
Etudier les principaux polymorphismes génétiques et leur impact sur la réponse thérapeutique aux médicaments utilisés en oncologie, en cardiologie, en neuropsychiatrie et dans la transplantation d’organes.
Etudier la corrélation entre le profil génétique des patients et la pharmacocinétique des médicaments.
Etudier la corrélation entre la réponse clinico-biologique (efficacité/toxicité) et le profil pharmacogénétique.
Concevoir des modèles de pharmacocinétique de population à partir de données de pharmacogénétique et de pharmacocinétique identifiées pour une meilleure personnalisation de la prescription avec une prédiction des doses afin d’optimiser les traitements médicamenteux, tant en termes d’efficacité que de sécurité d’emploi.
Créer une bio banque et d’une banque de données personnelles, pharmacogénétiques et pharmacocinétiques.
Etablir des recommandations pour l’indication de l’étude pharmacogénétique en fonction des modèles conçus.
Fiches de renseignements
Equipe

Pr. Sameh TRABELSI
Chef de service

Pr. Issam SALOUAGE
PROF

Pr. Emna GAIES
PR.AG

Pr. Rym CHARFI
PR.AG

Dr. Mouna BEN SASSI
AHU

Dr. Nadia JEBABLI
MAU

Dr. Hanene EL JEBARI
A.U
Dr. Syrine Ben-Hammamia
AHU
Galerie de photos